The “hunt” for an answer continues
Functional deficits have been identified in striatal neurons of mice expressing the mutation for Huntington’s disease. This research identifies a potential target for therapy treatment.
Author: Shelby Brock
Download: [ PDF ]
Neurophysiology
Huntington’s disease (HD) is a terminal neurodegenerative disorder which is the result of a repeating sequence of three nucleotides, CAG, in the huntingtin (HTT) gene.1 Repeats in this sequence are normal, but when it is repeated more than 40 times, the carrier is almost guaranteed to develop HD. Symptoms begin with difficulty concentrating and tremors and progress to involuntary movement (called chorea) and psychiatric decline.2 This is an autosomal dominant disease, meaning that the offspring of someone with Huntington’s has a 50% chance of developing the disease themselves. While there is no cure for this disease, most therapies focus on the treatment of symptoms.
In the pursuit of mechanistic knowledge of this disease, McAdam et al.’s team at the University of Edinburgh published a paper in Neurobiology of Disease which focused on the striatal neurons of mice with and without the HTT mutation.3 The striatum was chosen because it is rich in excitatory medium spiny neurons (MSNs), which are known to cause chorea (uncontrollable movement) when degenerated because the striatum is associated with voluntary movement.4 The researchers chose to study cell properties during high and low stimulation to identify latencies or defects. They hypothesized that MSNs are vulnerable to presynaptic dysfunction because they receive high amounts of excitatory input but would be unable to sustain those inputs overtime.
In the study, three groups of mice were used: wild type, homozygous HTTQ140/Q140 mutants, and heterozygous HTTQ140/+ mutants. The mutant mice have human mutations of the CAG repeat inserted into their genome, making them “knock-in” mice. A culture of striatum neurons was made from each mouse and the firing properties were assessed.
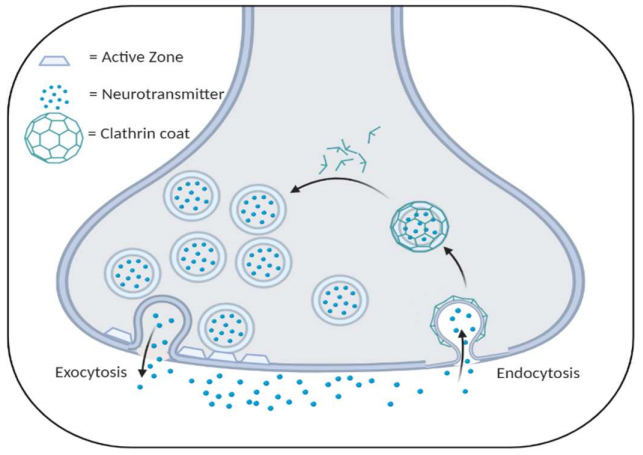
Normal synapses have synaptic vesicles which release neurotransmitter into the synaptic cleft (exocytosis) and are taken back into the presynaptic cell (endocytosis) to be recycled into the vesicle pool to be released again. This process is illustrated in Figure 1. McAdam’s team found that the cells of the homo- and heterozygous mice had significantly lower amounts of vesicle endocytosis, which would result in lower vesicle availability, lower neurotransmitter release, and, eventually, synaptic failure because of an inability to sustain the high volume of input being relayed through the striatum. However, this discrepancy in vesicle fusion timing only occurred at high-frequency stimulation. This coincides with the author’s hypothesis that synaptic dysfunction stems from an inability to maintain high levels of input.
Activity patterns stimulate the release of dopamine from medium spiny neurons, which is responsible for expected reward behavior and smooth coordination of movement. Some medium spiny neuron function relies on constantly active neurons to temporarily cease firing to encode salient environmental cues.5 If these cells do not fire consistently or cease firing at the wrong time, then the information input from other regions cannot be properly encoded, which includes voluntary movement.
Since this study’s results were found in both the homo- and heterozygous mice before physical symptoms of HD occurred, this suggests that those with the mutation have inherent dysfunction of their cells for their entire life. Slowed vesicle endocytosis may serve as a possible mechanism that might compound over time to promote the full manifestation of HD. Since this identifies a discrepancy in function before degeneration begins, it can be inferred that it is involved with the pathology of HD.
These findings could potentially stem future research on how vesicle timing leads to degeneration and potentially target the striatal excitatory neurons (specifically, vesicle fusion) when considering pharmacological ways to slow the progression of Huntington’s. Future experiments should be conducted in vivo to verify results and test how vesicle fusion changes over time with full inputs from other brain regions.
[+] References
Bates, G. The molecular genetics of Huntington disease — a history. Nat Rev Genet 6, 766–773 (2005). https://doi.org/10.1038/nrg1686.
Huntington's disease. (2020, April 14). Retrieved from https://www.mayoclinic.org/diseases-conditions/huntingtonsdisease/symptoms-causes/syc-20356117.
Mcadam, R. L., Morton, A., Gordon, S. L., Alterman, J. F., Khvorova, A., Cousin, M. A., & Smillie, K. J. (2020). Loss of huntingtin function slows synaptic vesicle endocytosis in striatal neurons from the httQ140/Q140 mouse model of Huntingtons disease. Neurobiology of Disease, 134, 104637. doi: 10.1016/j.nbd.2019.104637.
Dingman, M. (2015, February 16). Know your brain: Striatum. Neuroscientifically Challenged. Retrieved from https://www.neuroscientificallychallenged.com/blog/know-your-brain-striatum
Kim, T., Capps, R. A., Hamade, K. C., Barnett, W. H., Todorov, D. I., Latash, E. M., … Molkov, Y. I. (2019). The Functional Role of Striatal Cholinergic Interneurons in Reinforcement Learning From Computational Perspective. Frontiers in Neural Circuits, 13. doi: 10.3389/fncir.2019.00010
[+] Other Work By Shelby Brock
Dopamine receptors aren’t always dope
Neuroanatomy
The presence of specific allelic variants of the polyamorous dopamine receptor D4 (DRD4) is shown to be predictors of intensified neurodegeneration in frontotemporal dementia-spectrum patients.
Effects of neonatal methamphetamine exposure
Neuroscience In Review