The interactive role of Aβ plaques and tau pathology and their effects in Alzheimer’s disease
Author: Petru Buracioc
Download: [ PDF ]
Neuroscience In Review
Introduction
Alzheimer’s disease (AD) is a neurological disorder that affects nearly 50 million people worldwide.1 This disease is the leading cause of dementia which is characterized by cognitive impairment and memory loss.2 Individuals with AD suffer from behavioral changes and inability to recognize familiar faces as a result of neurodegeneration. In the United States, AD affects over 5 million adults and it is expected to increase up to 14 million by 2050 (CDC). The complexity of this disorder has made it difficult for therapeutic drugs to successfully eliminate the symptoms of AD or slow down its progression.
AD is characterized by extracellular amyloid-β (Aβ) plaques and intracellular tau tangles but the relationship between the two pathologies are poorly understood.3 Aβ plaques develop early during AD development and tau aggregates later in the stages of the AD. The amyloid cascade hypothesis suggests that the deposition of amyloid-β peptide in the brain sets up a series of pathological events that lead to neurodegeneration and Alzheimer’s disease. Such series of events include the possibility of Aβ plaques facilitating the aggregation of tau tangles which eventually leads to neuronal loss and cognitive impairment in AD. The amyloid cascade hypothesis has not been proven by studies examining the interaction between the Aβ plaques and tau pathologies using transgenic mice overexpressing human tau.4 Also, the accumulation of Aβ in the brain is poorly related to cognitive decline in AD patients, therefore the amyloid cascade hypothesis that can facilitate the over phosphorylation of tau should be considered. However, evidence does suggest that genetic mutations in amyloid precursor protein are associated with AD that can trigger other pathologies such as tau tangles and cholinergic dysfunction. A recent study found that Aβ plaques do in fact facilitate an environment that induces the formation of tau aggregates initially detected as tau aggregates in dystrophic neurites followed by the formation of neurofibrillary tangles.5 This finding is a breakthrough in AD research as it relates the two main contributors of AD by understanding the progression and timeline of AD, early treatments can be developed targeting different stages of AD and potentially slow down the progression of AD and increase the age of symptom onset in AD patients.
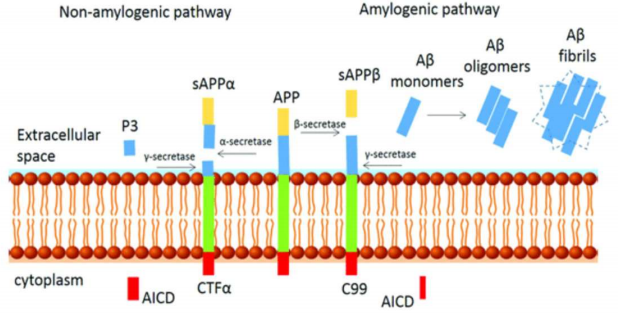
The reduction of Aβ accumulation in the brain is the center and the ultimate target of the amyloid hypothesis. Several antibodies that targeted regulators of APP proteolysis to block the β- or γsecretase pathways or increase α-secretase activity to reduced Aβ production (figure 1) were tested heavily in clinical trials. These studies failed to improve cognition in AD patients and were terminated. Also, reduction of over phosphorylated tau protein (tau tangles) has been successful with protein capped cadmium nanoparticles.6,7 Understanding the mechanism of AD such as the interaction between Aβ and tau pathologies can aid in development of drugs that will target early stages of AD.
The amyloid cascade hypothesis and its effects on the brain
The amyloid hypothesis received substantial attention from the academic community and pharmaceutical industries for the past two decades.1 Mounting evidence indicates that Aβ accumulation plays a key role in AD progression. For instance, the β-secretase enzyme which is also called β-site APP cleaving enzyme I (BACE1) has been linked extensively to AD as it initiates the cleavage of APP generating abnormal Aβ fibrils. β-secretase activity is present throughout the body but significant increase in β-secretase activity was found in neurons of AD patients.8 BACE1 knockouts studies showed that blocking BACE1 activity in APPoverexpressing transgenic mice diminished amyloid deposition significantly, suggesting that BACE1 is essential to amyloid formation.9 However, more recent studies found that complete deletion of BACE1 in mice showed adverse effects such as impaired spatial reference and working memories.10 One BACE1 inhibitor called Verubecestat was tested in AD patients and due to its adverse effects, such as aggravated cognition and was terminated in phase III clinical trials.11 These studies confirm that complete obliteration of BACE1 and thus complete abolishment of Aβ has adverse effects in AD patients. Additionally, BACE1 knockout mice suggests that normal levels of Aβ is vital to normal functioning of the brain such as memory performance.12 Another therapeutic that attempted to lower Aβ accumulation was a γ-secretase inhibitor called Semagacestat. This inhibitor reduced amyloid deposition but increased cognitive impairment in AD patients and its use was halted.13 These results are in conjunction with other studies that showed Aβ is vital to physiological function and may be involved in neuronal growth and memory.14 The failure to produce potent and efficacious therapeutics by targeting the inhibition of Aβ accumulation in AD suggests that therapeutics can be efficacious only when the Aβ accumulation is reduced to a critical level and not completely eliminated.
Aβ accumulation inducing tau pathology in AD transgenic mouse models
AD is characterized by both extracellular amyloid-β (Aβ) plaques and intracellular tau tangles but the relationship between the two pathologies are poorly understood. Aβ plaques develop early during AD development in the cortex and hippocampus while tau over phosphorylation takes place later in the stages of AD.5,15 Initially, tau aggregates were found to form in the transentorhinal cortex and further spread to the hippocampal formation and neocortex.16 Evidence supporting the missing interaction between amyloid-β and tau tangles is the tau mutation in chromosome 17 that causes autosomal dominant frontotemporal lobe dementia which is not accompanied with amyloid-β plaques.17 However, this claim is insufficient to deny the amyloid cascade hypothesis because studies using transgenic mice models suggest the interaction with Aβ and tau leads to the progression of AD.
A study crossed JNPL3 transgenic mice expressing a mutant tau protein with Tg2576 transgenic mice expressing mutant ß-amyloid precursor protein (APP). When compared to the mice that only expressed mutant tau protein, the double mutant mice showed significant increase in AD-like neurofibrillary tangles (NFTs) in the limbic system and olfactory cortex.18 Another study inserted Aß fibrils into JNPL3 transgenic mice expressing a mutant tau protein and determined that Aß induced NFTs formation as fast as 18 days after the injection. Another similar study generated a triple transgenic model of AD (3xTg-AD) harboring three AD mutant genes: APP, preselin 1, and tauP301.19 In this study amyloid-β deposition was identified prior to the expression of NFTs supporting the amyloid cascade hypothesis that beta amyloid facilitates formation of NFTs and other tau pathologies. A more recent study injected human derived tau fibrils into the hippocampus and the cortex of mice with knock in mutated APP genes.5 The study found that amyloid-β plaques do in fact facilitate an environment that promotes the development of tau tangles. These tangles initially form as tau aggregates in dystrophic neurites surrounding Aβ plaques which then spread throughout the brain.5 Tau pathology is considered a downstream event in the amyloid hypothesis suggesting that it is responsible for significant neuronal damage and cognitive impairment.13
Conclusion
The amyloid cascade hypothesis is still debatable in AD research in part due to mounting evidence that suggests that amyloid beta is not entirely responsible for the progression of AD. Studies show that some individuals with elevated beta-amyloid accumulation do not suffer from impaired memory and learning. However, it is evident that mutations in the gene that codes for this protein might trigger other pathological events in AD patients that further damage the brain. Also, the inability to generate efficacious and potent drugs that can reduce amyloid accumulation without adverse effects is considered evidence against this hypothesis. Multiple studies using sophisticated mouse models with more than one gene mutation suggest that this hypothesis may induce tau pathology and causes neural loss. On the other hand, evidence suggesting that there is no link between amyloid deposits and tau pathology is insufficient and lacks physiological studies. Expansion of existing research generated from 25 years of amyloid focused research should further investigate the interaction between amyloid deposition and tau pathology utilizing existing successful transgenic mice models. First and foremost, the transgenic mouse models used in studying AD pathologies need to be reexamined as failed clinical trials suggested that AD pathologies represented in humans differ from these mouse models where drugs that seemed to significantly increase cognition in mice did not improve cognition in AD patients. One question that has not been answered by amyloid AD research is where in the when in the stage of AD would the amyloid-β-directed therapeutic be most effective? To answer this question, studies need to investigate when in the stage of AD would the amyloid-β-directed therapeutic be most effective? Besides the interaction of amyloid with tau pathology, studies should investigate the interaction of the amyloid hypothesis with the cholinergic hypothesis as research suggests these two pathologies interact in different nicotinic acetylcholine receptors which causes cognitive impairment. Such evidence include agonists and positive allosteric modulators on nicotinic receptors alleviate cognitive impairments in mouse models.
[+] References
Sumner, I. L., Edwards, R. A., Asuni, A. A., & Teeling, J. L. (2018). Antibody Engineering for Optimized Immunotherapy in Alzheimer's Disease. Frontiers in neuroscience, 12, 254. https://doi.org/10.3389/fnins.2018.00254
Teunissen, C. E., De Vente, J., Steinbusch, H. W. M., & De Bruijn, C. (2002). Biochemical markers related to Alzheimer’s dementia in serum and cerebrospinal fluid. Neurobiology of aging, 23(4), 485-508
Forman MS, Trojanowski JQ, Lee VM. Neurodegenerative diseases: a decade of discoverie paves the way for therapeutic breakthroughs. Nat Med. 2004;10(10):1055–1063. doi:10.1038/nm1113
Ricciarelli, R., & Fedele, E. (2017). The Amyloid Cascade Hypothesis in Alzheimer's Disease: It's Time to Change Our Mind. Current neuropharmacology, 15(6), 926–935. https://doi.org/10.2174/1570159X15666170116143743
He, Z., Guo, J., McBride, J. et al. Amyloid-β plaques enhance Alzheimer's brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat Med 24, 29–38 (2018) doi:10.1038/nm.4443
Leslie Crews, Eliezer Masliah, Molecular mechanisms of neurodegeneration in Alzheimer's disease, Human Molecular Genetics, Volume 19, Issue R1, 15 April 2010, Pages R12– R20, https://doi.org/10.1093/hmg/ddq160
Shweta Kishor Sonawane, Absar Ahmad, and Subashchandrabose Chinnathambi, ACS Omega 2019 4 (7), 12833- 12840 DOI: 10.1021/acsomega.9b01411
Seubert P, Oltersdorf T, Lee MG, Barbour R, Blomqist C, Davis DL, Bryant K, Fritz LC, Galasko D, Thal LJ, Lieberburg I, Schenk DB: Secretion of b-amyloid precursor protein cleaved at the amino-terminus of the b-amyloid peptide. Nature. 1993, 361: 260-263.10.1038/361260a0.v
Laird FM, Cai H, Savonenko AV, Farah MH, He K, Melnikova T, Wen H, Chiang HC, Xu G, Koliatsos VE, Borchelt DR, Price DL, Lee HK, Wong PC: BACE1, a major determinant of selective vulnerability of the brain to amyloid-beta amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J Neurosci. 2005, 25 (50): 11693- 11709. 10.1523/JNEUROSCI.2766-05.2005
Cole, S.L., Vassar, R. The Alzheimer's disease β-secretase enzyme, BACE1. Mol Neurodegeneration 2, 22 (2007). https://doi.org/10.1186/1750-1326-2-22
Egan, M.F., Mukai, Y., Voss, T. et al. Further analyses of the safety of verubecestat in the phase 3 EPOCH trial of mild-to-moderate Alzheimer’s disease. Alz Res Therapy 11, 68 (2019). https://doi.org/10.1186/s13195-019-0520-1
Ohno M, Sametsky EA, Younkin LH, Oakley H, Younkin SG, Citron M, Vassar R, Disterhoft JF: BACE1 Deficiency Rescues Memory Deficits and Cholinergic Dysfunction in a Mouse Model of Alzheimer's Disease. Neuron. 2004, 41 (1): 27-33. 10.1016/S0896-6273(03)00810-9.
Karran, E., Mercken, M. & Strooper, B. The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov 10, 698–712 (2011). https://doi.org/10.1038/nrd3505
Pearson, H. A., & Peers, C. (2006). Physiological roles for amyloid beta peptides. The Journal of physiology, 575(Pt 1), 5–10. https://doi.org/10.1113/jphysiol.2006.111203
Hsiao, K., Chapman, P., Nilsen, S., Eckman, C., Harigaya, Y., Younkin, S., . . . Cole, G. (1996). Correlative Memory Deficits, A Elevation, and Amyloid Plaques in Transgenic Mice. Science, 274(5284), 99-103. doi:10.1126/science.274.5284.99
Braak, H., & Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathologica, 82(4), 239-259. doi:10.1007/bf00308809
Hutton, M. et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393, 702–705 (1998)
Lewis, J., Dickson, D. W., Lin, W. L., Chisholm, L., Corral, A., Jones, G., ... & Eckman, C. (2001). Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science, 293(5534), 1487-1491
Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer's disease. Neurobiol Aging. 2003;24:1063–1070
[+] Other Work By Petru Buracioc
Hope for schizophrenia patients after discovery of brain cells associated with this mental chronic disorder
Neuroanatomy
There are limited studies underlying the basis of schizophrenia that can be used to model experiments to expand the genomic basis of schizophrenia and help alleviate its symptoms more effectively then the treatments available today. That is exactly what a recent study has discovered. This study incorporated knowledge of the brain from single-cell RNA sequencing and analyzed whether specific loci associated with schizophrenia were mapped on specific cells in the brain.
A new drug approach increases cognition in rats
Neurophysiology
In a new study the α4β2 nicotinic acetylcholine receptors, a subtype of nicotinic acetylcholine receptors was studied in relation to their role in attention and cognition. This research presents a novel approach using a positive allosteric modulator on α4β2 nicotinic acetylcholine receptors to increase cognition in rats. This approach can be used in cognitive deficit disorders such as Alzheimer’s Disease and schizophrenia, which show a reduction of α4β2 nicotinic acetylcholine receptors in several parts of the brain.